
Spinal muscular atrophy (SMA) is a rare but serious neuromuscular disorder that causes worsening muscle weakness. SMA is a genetic, or inherited, disorder caused by a change in the SMN1 gene that leads to insufficient levels of a critical protein called survival motor neuron (SMN). As a result, the nerve cells that signal muscles to contract (motor neurons) degenerate, contributing to progressive muscle weakness, difficulty breathing and swallowing, and, in severe cases, early death.
Currently, there is no cure for SMA. Treatment is primarily aimed at managing symptoms and preventing complications. Current medications aim to stimulate the production of the SMN protein to support motor neuron survival. However, patient response suggests these therapies may not restore motor function to normal levels, with motor function plateauing or continuing to decline. There is a high need for therapeutic approaches that preserve muscle strength.
A recent study led by Stanford Cardiovascular Institute researchers, published in Circulation Research, focuses on the role of skeletal muscle in SMA and proposes a novel combination therapy approach that directly targets diseased muscle cells.
Building a Patient-specific Model of SMA
The team was led by first authors Wenshu Zeng and Xiaohui Kong and senior authors John Day (Director, Neuromuscular Division and Clinics) and Joseph Wu (Director, Stanford Cardiovascular Institute). They used human induced pluripotent stem cells (iPSCs) to create a model system of SMA. iPSCs are a powerful tool that allows researchers to turn cells obtained from patients or healthy control donors into stem cells that are then differentiated into various cell types. Importantly, these iPSCs retain the genetic makeup of the donor so they can be a personalized platform for studying rare diseases, such as SMA.
In this study, the team generated iPSC derived-skeletal muscle cells (iPSC-SKMs) from patients with SMA Type 1, the most severe form of the disease, and compared them to iPSC-SKMs from healthy individuals. Importantly, they found that the SMA iPSC-SKMs could model key aspects of the disease, such as a lower SMN protein levels and reduced size of muscle fibers and , making them an ideal platform for studying SMA.
Uncovering a Source of Cellular Stress in SMA Muscle
The iPSC-SKM platform allowed the team to better understand why SMA muscle cells fail to properly develop. Using a global protein analysis, they identified proteins that were either increased or decreased in SMA cells compared to controls. They identified proteins needed for muscle contraction and structure were significantly reduced in SMA cells. A notable finding was a robust increase in reactive oxygen species (ROS), molecules that can damage cells when present at a high level. ROS are often a sign of oxidative stress, which is also known to worsen symptoms of muscular disorders. Interestingly, the team also identified monoamine oxidase A (MAOA) as a key contributor to the elevated ROS, and higher MAOA levels were present in SMA muscle cells.
A Promising Therapy
Learning that MAOA may actively contribute to muscle damage in SMA makes it a potential therapeutic target. Genistein is a naturally occurring compound found in soy that is used for various health conditions. It is also known to inhibit MAOA activity. The Stanford team showed that genistein could reduce MAOA activity and lower ROS levels in SMA muscle cells, as well as improve muscle fiber size. This indicates an intriguing benefit to muscle health.
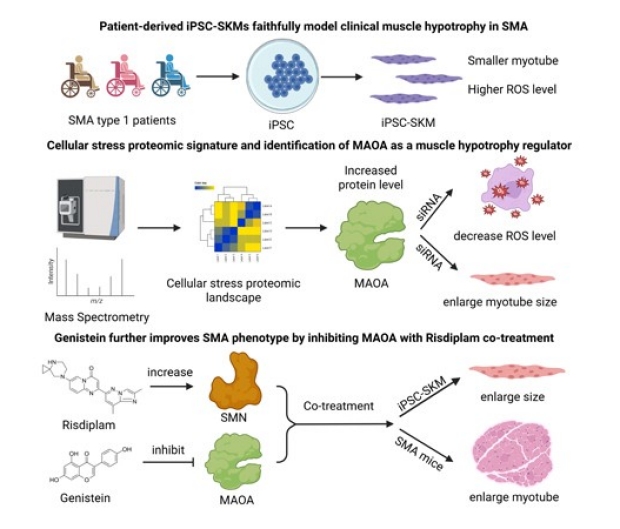
Patient-derived iPSC skeletal muscles (iPSC-SKMs) serve as an ideal platform to study SMA and discover potential therapies. Combining risdiplam and genistein, a SMN-enhancing drug and a MAOA inhibitor, has the potential to improve outcomes more than either treatment alone, as seen in both iPSC-SKMs and SMA mice.
The team also tested genistein along with risdiplam, an FDA-approved drug that increases SMN protein levels. Alone, each drug had a modest effect. In combination, there was a stronger effect, with reduced oxidative stress and improved muscle fiber size. The team also tested the combination therapy in a mouse model of SMA. When genistein and risdiplam are combined, the treatment enhanced muscle fiber size in vivo.
This study offers several important insights into SMA. They found that SMA muscle dysfunction is partially due to the muscle cells themselves, rather than just the nerve input they receive. They showed that MAOA as a novel target for reducing oxidative stress and improving muscle health in SMA. Significantly, they also show that combining risdiplam and genistein, a SMN-enhancing drug and a MAOA inhibitor, has the potential to improve outcomes more than either treatment alone. These critical findings underscore the promise if iPSC-based disease modeling for rare diseases.
Additional authors include Arianne Caudal, Shane Zhao, Christina Alamana, Juan Melesio, and Lu Ren from the Stanford Cardiovascular Institute; Jessica Guzman from Department of Neurology; Kelly Howell and Karen Chen from Spinal Muscular Atrophy Foundation; and Paul Pang from Greenstone Biosciences. The study was supported by the National Institutes of Health, Spinal Muscular Atrophy Foundation, American Heart Association, and the Stanford Maternal and Child Health Research Institute.