
Formalin-fixed, paraffin-embedded (FFPE) tissues represent the predominant sample conservation method in clinical practice, yet degraded and crosslinked RNA has long limited whole-transcriptome analysis and spatial context.
On August 28, in Cell, researchers at BGI-Research and collaborating clinical and research centers published Stereo-seq V2, a spatial transcriptomics method that achieves single-cell–level resolution on FFPE sections and, critically, enables simultaneous in situ profiling of host and microbial RNAs. By combining deparaffinization/decrosslinking with random-primed capture and uniform gene-body coverage, Stereo-seq V2 opens FFPE "black boxes" for infectious disease biology, tumor ecosystems, and clinical research applications.
The study “Stereo-seq V2: Spatial
mapping of total RNA on FFPE sections with high resolution” was published in
Cell.
Built
on the large field of view and subcellular spot spacing of Stereo-seq, the V2
chemistry replaces poly(T) capture with random primers, increasing sensitivity
to low-abundance and non-poly(A) RNAs while maintaining signal-to-noise. This
advance enables sharper boundaries and cellular resolution, with V2 reducing
lateral diffusion at tissue borders and achieving true single-cell segmentation on FFPE sections, identifying 34 cell
types in mouse brain. Across adjacent mouse brain sections, V2 and V1 showed
high concordance in gene expression and reduced apparent lateral diffusion at
anatomical borders. Compared
with existing probe-based methods, V2 provides whole-transcriptome coverage,
significantly expanding beyond traditional probe limitations, while recovering
important markers and regulatory genes otherwise missed or inconsistently
captured by probe panels. Uniform coverage across exons improves detection of
transcription factors, adhesion genes, and alternative splicing events,
categories that often prove elusive with 3′-biased strategies.
Demonstrating
clinical FFPE robustness, V2 maintains high gene capture even on severely
degraded samples and aligns molecular signatures with histological features
like necrosis. V2 performs robustly on clinical FFPE samples with poor RNA
integrity. In a cohort of triple-negative breast cancer (TNBC) blocks preserved
from under 1 to ~9 years, V2 maintained consistent gene capture. The method
showed reliable performance across different tissue conditions. Spatial
expression recapitulated clinical immunohistochemical phenotypes (e.g., low
ESR1/PGR/ERBB2), delineated malignant, immune, and necrotic regions, and
supported tumor subtype classification through genetic analysis. Whole
gene-body coverage enabled spatial alternative splicing analysis, identifying
1,492 events including specific splicing patterns in tumor-associated genes
like ZNF226, GIPC1, and BEX4 that varied between tumor subtypes. These findings
add molecular granularity to tumor biology beyond expression alone.
A
core advantage is unbiased, concurrent
mapping of host and microbial RNAs directly on FFPE tissue, enabling
host–microbe battlefield mapping where V2 simultaneously maps spatial B-cell
receptor (BCR) clones in human Tuberculosis (TB)
lungs and tracks Mtb infection dynamics with bacterial RNA peaking at 4 weeks
in mouse models. In
a Mycobacterium tuberculosis (Mtb) mouse model profiled at 1 day, 4 weeks, and
8 weeks post infection, V2 captured the rise-and-fall kinetics of bacterial
transcripts consistent with histology and CFU counts and identified host gene
modules spatially correlated with Mtb burden, shifting from inflammatory and
cell death pathways to adaptive immunity over time. Stereo-seq
V2 further resolves the in situ BCR repertoire. Compared with
poly(A)-based methods, V2 provides markedly improved coverage of V regions,
enabling assembly of clonotypes and analysis of somatic hypermutation. The
number of assembled BCR clones increased from 185 at 4 weeks to 1,736 at 8 weeks,
with greater clonal diversity and mutation frequency proximal to infected
regions, consistent with affinity maturation. In
human TB FFPE lung samples, V2 similarly mapped host and pathogen RNAs,
delineated necrotic boundaries from RNA density, and revealed recurrent BCR
clones across patients; related clones were enriched in blood from active TB
cases, pointing to potential diagnostic and therapeutic targets.
Together,
these results position Stereo-seq V2 as a broadly applicable platform for
single-cell–level, whole-transcriptome mapping on FFPE sections, extending
spatial transcriptomics to the most abundant and clinically relevant specimens.
By unifying host gene programs, immune repertoires, and pathogen localization
within the same section, V2 provides a panoramic readout of tissue ecosystems
in cancer, infection, and beyond, and may accelerate retrospective biomarker
discovery, mechanism-based stratification, and antibody or vaccine development
using existing archives.
All
human and animal studies were conducted with appropriate institutional ethics
approvals and followed established guidelines for research involving clinical
specimens and laboratory animals. Data and code availability: Raw data are
deposited in the Genome Sequence Archive; analysis code is available at the
project repository: GitHub (Stereo-seq V2 code) at https://github.com/YoungLi88/Stereo-seq-V2. The study can be accessed here: https://www.cell.com/cell/fulltext/S0092-8674(25)00922-5?rss=yes
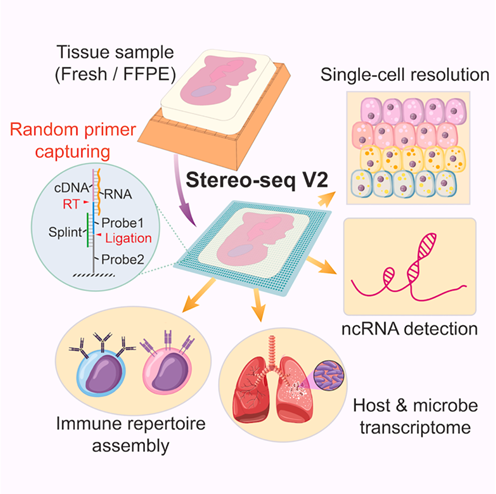
Stereo-seq V2 workflow enables simultaneous host and microbial RNA mapping
on FFPE tissues with single-cell resolution.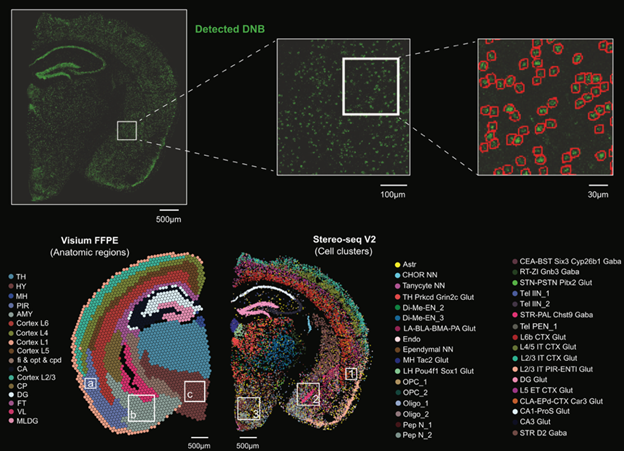
Stereo-seq V2 demonstrates improved spatial resolution with reduced lateral
diffusion at tissue boundaries and achieves single-cell resolution on FFPE
sections, identifying 34 distinct cell types in mouse brain.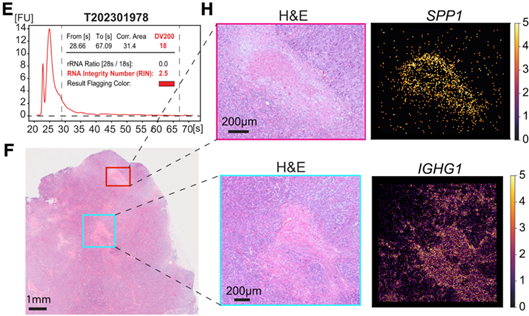
V2 maintains robust gene capture across degraded clinical TNBC samples and
accurately maps tumor, immune, and necrotic regions.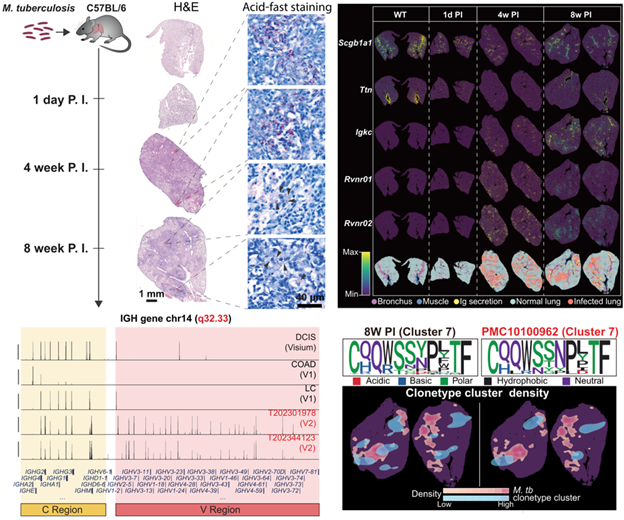
Spatial BCR repertoire analysis reveals clonal expansion and affinity
maturation in TB-infected tissues, with related clones detected in patient
blood.